
抗体偶联药物(ADC)的设计需要综合考虑抗体 、 连接子 、 小分子毒素三个组成成分及它们之间的合理组合 。 其中抗体的选择是 ADC 设计的起点 , 连接子与连接技术是决定 ADC 成药性的关键因素 , 小分子药物对于肿瘤细胞应具有高效的杀伤作用 。 ADC 药物的设计也一直围绕着这三部分不断发展 。ADC 药物的工艺包括了小分子的制备和抗体的制备 , ADC 分析也相较于传统生物制药分析更为复杂 , 需要多种生物制药研究和分析方法联合使用。
今天为大家解读一篇关于ADC药物的CMC与表征概述的文章。主要围绕第一代ADC药物进行阐述,通过了解第一代ADC的开发可以让我们站在巨人的肩膀上为新一代ADC研发提供思路。

ADC从工艺和分析开发的角度来看,其技术体系极为复杂。ADC通过规避细胞毒性的方式实现高效活性成分的靶向递送,相比其单个分子组分能提供更优越的治疗指数,正成为该领域新兴的解决方案。ADC产品的质量与性能直接取决于生产工艺及其设计控制方式。本章将重点阐述第一代ADC的工艺开发,以及这些工艺在新一代ADC中的延伸应用。
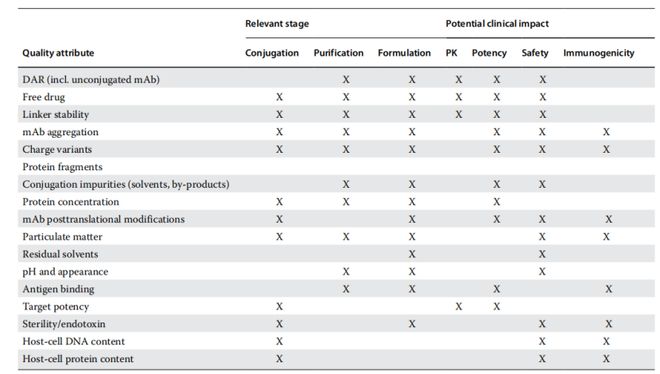
ADC的开发从充分表征的单克隆抗体(mAb)、连接子及细胞毒素原料开始,依次经历mAb活化、偶联、纯化、制剂及临床给药储存等步骤。图1.1概括了第一代ADC产品的通用生产工艺流程。在整个过程中,ADC各组分(抗体、毒素和连接子)均会经历严苛的化学与物理环境(条件),这些条件有可能影响产品质量,应进行充分的控制。关键质量属性(CQAs,见表1.1)是ADC工艺的核心概念,需在整个流程中进行全面监控以确保终产品的高质量。监控CQAs的目的是根据质量目标产品概况(QTPP)要求——包括安全性和有效性等特性——确保产品达到既定质量标准。
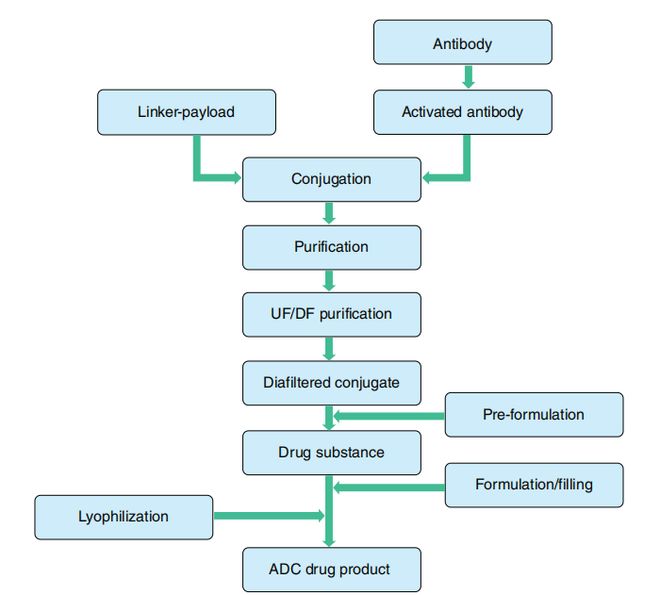
ADC的生产始于化学与生物起始原料,首先抗体与小分子偶联生成ADC原液。从工艺规模来看,正是这一偶联工艺环节使ADC产品开发既不同于单抗药物,也区别于小分子药物开发。该步骤需在支持活性化学反应条件下进行,既要实现单抗的适度偶联,又要避免起始原料发生显著降解或交叉反应。
在进入偶联阶段前,ADC工艺中的连接子、细胞毒素有效载荷和单抗中间体的生产与控制要求存在显著差异。此外,完成整个ADC制剂(DP)生产所需的设施需具备细胞毒素处理、活性化学反应、生物制品及高分子量蛋白质操作等综合能力。
理想情况下,偶联工艺及后续纯化、制剂过程不应导致单抗与细胞毒素中间体产生新的降解或不稳定途径。但事实上,相较于亲本单抗,这一新分子实体本身稳定性降低,因此必须采取相应措施以最大限度减少这些不稳定因素。
第一代ADC产品(Kadcyla®和Adcetris®)分别代表了基于赖氨酸和基于半胱氨酸的偶联策略。Kadcyla®采用双功能连接子SMCC(N-琥珀酰亚胺基-4-(马来酰亚胺甲基)环己烷-1-羧酸酯):首先其琥珀酰亚胺基团与曲妥珠单抗的多个赖氨酸侧链反应生成活化抗体,随后SMCC连接子的马来酰亚胺端基与DM1毒素上引入的巯基反应,最终形成完整偶联物。
以Adcetris®为例的基于巯基的偶联工艺中,首先采用三(2-羧乙基)膦(TCEP)对链内二硫键进行可控还原,在抗体活化阶段产生游离巯基。随后通过马来酰亚胺功能化的连接子-载荷复合物(含蛋白酶可切割的缬氨酸-瓜氨酸连接子及MMAE毒素)对这些游离巯基进行标记。与赖氨酸偶联相比,半胱氨酸偶联产生的偶联产物分布更为均一。
在赖氨酸与半胱氨酸偶联过程中,均可能发生多种副反应。琥珀酰亚胺或马来酰亚胺的反应活性在中性/碱性pH条件下增强,但该环境同时会促进单抗发生脱酰胺反应、焦谷氨酸形成及蛋白质聚集。偶联工艺需避免亲核试剂干扰,并通过精确的pH控制平衡化学反应活性与单抗稳定性。
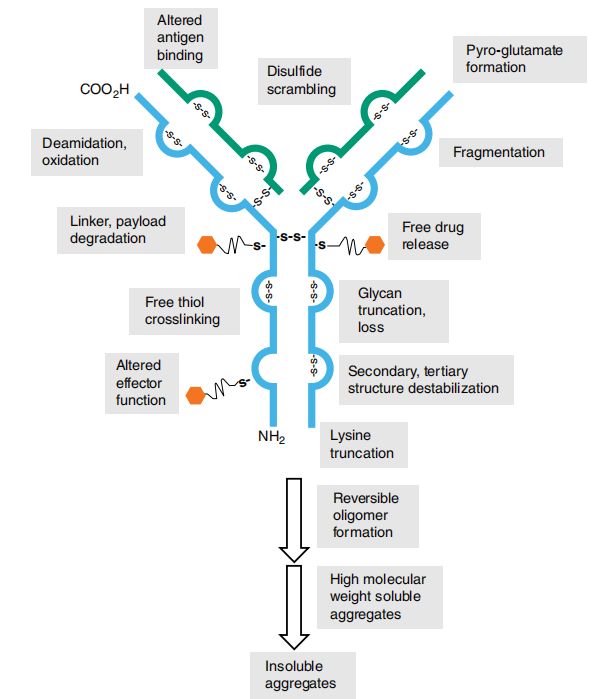
偶联异质性也可从竞争反应角度分析:赖氨酸或半胱氨酸侧链的可及性及反应活性,既受整体溶液条件影响,也取决于单抗二级/三级结构中的局部微环境。由于这些微环境会随单抗构象的细微变化而改变,单抗结构不稳定将直接影响偶联异质性和化学计量比。图1.2展示了ADC分子在制造过程中承受的化学与物理作用力——根据工艺要求,单抗分子在活化与偶联阶段需接触高温、有机溶剂、还原剂及反应副产物。此外,在抗体活化与偶联过程中还涉及缓冲液置换、浓度调节、色谱分离等多步中间操作。这些处理步骤虽然能实现ADC物质的富集或分离,但均可能引发物理不稳定性。
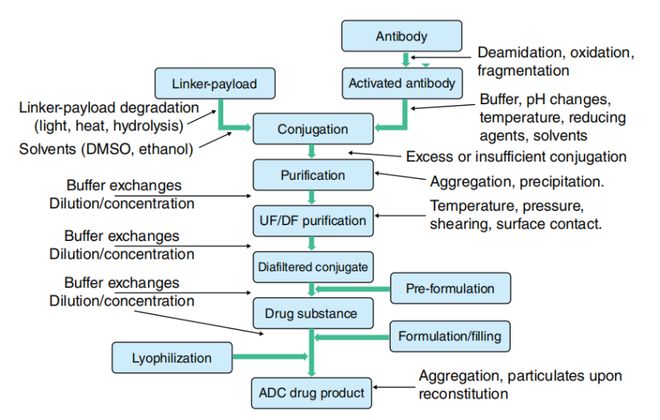
试剂的储存、操作与质量控制对实现稳定的偶联反应至关重要。以常用还原剂TCEP为例,其杂质含量或浓度偏差会导致游离巯基生成不足,进而降低偶联效率;连接子在运输或储存过程中若接触湿气,同样可能造成不完全偶联;而单抗的不当处理也会如前文所述影响偶联效果。偶联位点与化学计量比会显著影响ADC的疗效与安全性特征,因此必须对这些关键质量属性进行系统检测。
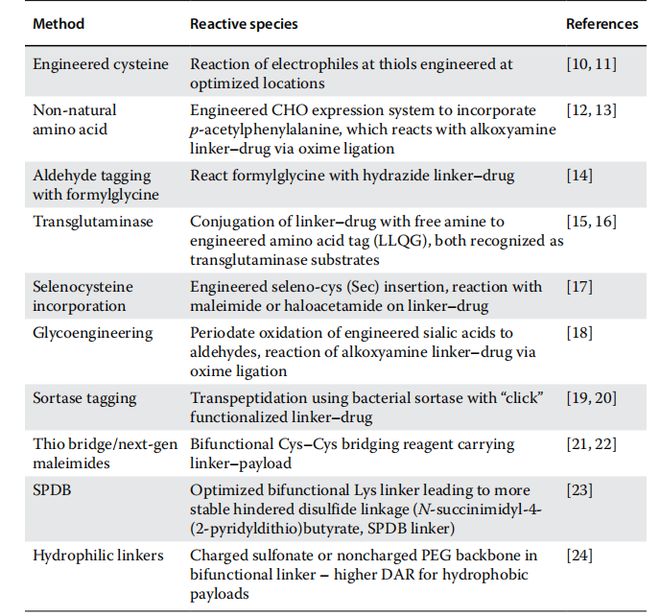
当前,新一代偶联化学技术的探索正在蓬勃开展,旨在拓展ADC的治疗应用范畴。这些突破或将深刻影响未来ADC的工艺发展方向。如表1.2所示,重点研发方向包括:新型有效载荷的开发、替代性连接子技术的创新,以及能够降低ADC异质性的优化生物偶联策略。
在下一代ADC设计中,目前主要有四类具有重要临床意义的细胞毒素——尽管这些毒素对整体ADC开发流程的影响差异不大。以Adcetris®(微管抑制剂奥瑞他汀类)和Kadcyla®(美登素类)为代表的微管蛋白抑制剂,以及基于DNA结合与嵌入机制的新型毒素。
例如从棘孢小单孢菌中分离的天然产物卡奇霉素(calicheamicin),可通过结合DNA小沟并引发链断裂发挥作用。这类抗肿瘤物质作为ADC细胞毒素已探索多年,首个获批ADC药物Mylotarg®(吉妥珠单抗奥佐米星)即采用该毒素。另一类吡咯并苯二氮卓类(PBDs)化合物同样通过DNA小沟结合与交联发挥作用。虽然这类分子效价极高,但其强疏水性和光敏感性也带来稳定性风险。还有阿霉素等。(第一代ADC载荷)
ADC连接子是一类多功能分子,既能确保ADC的稳定递送,又能实现细胞毒素载荷的靶向释放。从ADC工艺与质量控制的角度来看,连接子设计深刻影响着偶联反应条件及后续ADC产物的处理。这些分子既要提供连接细胞毒素与单抗的化学反应位点,又需具备可控释放载荷的功能结构——当ADC被内化后,其骨架结构能触发毒素释放。这些功能要求带来的技术挑战推动了该领域的广泛研究,并对ADC开发中的偶联及下游工艺产生深远影响。
ADC连接子本质上可分为不可切割型与可切割型两大类。相较于可切割连接子,Kadcyla®采用的SMCC不可切割型连接子,以及添加了聚乙二醇(PEG)间隔基,在工艺开发和特性分析方面难度较低。而可切割连接子则利用酸不稳定的腙键、可还原的二硫键或蛋白酶敏感的肽段(如缬氨酸-瓜氨酸)来实现细胞内毒素释放。
从工艺与生产稳定性考量,必须对这些试剂进行全程严格监控:包括偶联阶段前后的总纯度、微量降解物,以及可能影响稳定性的各种因素。理想情况下,连接子切割应仅发生在细胞内。但事实上,ADC原药在纯化、制剂和冻干等下游工艺中,始终暴露于pH变化、温度波动、光照和空气接触等外部压力,这些均可能导致游离药物微量释放,带来全程毒性风险。因此,所有下游生产环节都需采用全面且正交的分析方法,既要检测游离药物及其相关降解形式,更要监控ADC上连接子-载荷的脱落情况。
以常见的硫代琥珀酰亚胺键为例(通过硫醇与马来酰亚胺的迈克尔加成反应形成):该键可能通过琥珀酰亚胺水解或逆迈克尔反应发生降解。虽然开环水解产物不会导致载荷释放,但会改变ADC的溶液电荷状态;而逆迈克尔反应则会产生具有游离巯基反应活性的载荷形式,可能与其他可及巯基形成蛋白质-药物偶联物。这种连接子降解途径不仅会降低ADC生物活性,还可能引发毒性隐患。
基于硫醇或赖氨酸的连接子化学面临的核心工艺挑战在于偶联异质性,这种异质性是整个ADC工艺的关键控制点,需要建立全面的质量控制和深入的表征分析,以确保符合质量目标产品概况(QTPP)标准。以Kadcyla®为例,其曲妥珠单抗分子上最多可分布8个载荷,这一数量主要由SMCC连接子的活化步骤决定;而Adcetris®则通过TCEP控制二硫键还原程度来精准调控药物抗体比(DAR)。这两种工艺都强调对反应条件(浓度、pH、温度和时间)及起始物料质量的严格控制,这些因素直接影响产品放行必须达标的DAR值,同时还需避免单抗本身的过度降解或结构破坏。
若能获得更均一的ADC分子,将大幅简化从偶联到下游的整个生产工艺与表征流程。目前已有若干创新技术正在开发中,表1.2列举了部分代表性进展,这些技术有望解决第一代ADC面临的多种工艺挑战。其优势包括:降低DAR相关的偶联异质性、减少未偶联单抗残留、改善载荷位点分布——所有这些特性均已被证实会影响ADC的治疗窗和稳定性。此外,Fc功能与抗原结合亲和力、体内稳定性与药代动力学特性、以及内化后载荷释放效率等,也都与偶联程度和位点密切相关。
定点偶联策略主要包括:工程化半胱氨酸残基技术和通过优化表达系统引入非天然氨基酸。前者仍存在游离巯基引发的风险,可能形成分子内二硫键错配,若巯基位点选择不当会导致更严重的蛋白质结构变化。另一种定点偶联方法利用转谷氨酰胺酶(一种能催化谷氨酰胺与赖氨酸侧链成键的细菌酶,该酶可识别IgG恒定区工程化的谷氨酰胺标签(LLQG),通过酶法或更优的化学酶法策略可实现DAR值1-2的精确控制。
这些技术都不可避免需要使用活性化学或生物化学方法将小分子连接子与细胞毒素连接到单抗上。在纯化前的偶联步骤中,抗体和连接子-载荷复合物会经历多种严苛条件,包括pH波动(pH4-8)、高温以及共溶剂(如二甲基亚砜DMSO、乙醇)处理。从工艺开发角度看,始终存在副反应或降解风险,最终可能影响偶联收率。表1.2所述的若干新一代偶联技术采用了单抗一级结构修饰、改造表达系统或非天然氨基酸等策略,但工程化单抗的体外和体内稳定性可能不及现有上市抗体药物那样明确可靠。
在获得符合预期组分和活性的偶联抗体后,粗产物需经过纯化流程以去除大部分低分子量组分。根据偶联方案的不同,在抗体活化与偶联步骤之间可能还需进行中间纯化或缓冲液置换。ADC的纯化通常采用透析过滤技术——该技术体系通过使粗产物与特定截留分子量的超滤膜接触,在泵压驱动下使低分子量物质透过膜孔,而主要含ADC的高分子量物质则被截留。该工艺同时可实现ADC原液的缓冲液置换和浓度调整,使其更符合制剂要求。
透析过滤存在多种形式,尤其体现在规模差异上:小规模操作可通过离心或负压实现超滤,而工业化生产则采用切向流过滤(TFF)等连续流系统。虽然大规模TFF能为ADC原液提供更温和的处理环境,但空气接触、反复界面作用及温度波动等因素仍会带来稳定性风险(参见图1.2及表1.1/1.3),进而影响ADC关键质量属性。相较于未偶联的IgG分子,ADC因携带多个疏水性修饰基团而导致溶解性和稳定性下降,这要求其纯化过程需采取更专业的处理措施。
ADC制剂开发需实现双重目标:在初步纯化后产物稳定,并为后续冻干、复溶及静脉给药提供适宜载体。因此,制剂工艺是连接纯化ADC分子与临床使用产品的关键桥梁。在上市批准前,随着ADC产品标准的逐步确立,其制剂方案从临床前到III期研究阶段会持续优化。尽早明确给药途径对工艺开发至关重要——特别是在偶联阶段后,这将指导纯化与制剂策略的制定。制剂作为ADC整体生产工艺的核心环节,其典型组成包括缓冲剂、糖类和表面活性剂,这些组分共同维持ADC溶液的稳定性、冻干特性及复溶性能。
相较于单抗制剂,ADC制剂存在两个显著差异:其一,ADC浓度范围(Adcetris®:5mg/mL,Kadcyla®:20mg/mL)通常低于常规单抗制剂,因此对降低粘度的辅料需求减少;其二,连接子-载荷的化学稳定性成为缓冲剂选择和pH设定的关键考量因素。由于连接子-药物组分引入的额外疏水性,制剂开发中需重点监控聚集和构象不稳定性(参见图1.2及表1.3)。对此必须采用正交分析和物化检测技术,因为所观察到的降解或不稳定现象高度依赖于分析方法。
尽管存在这些差异,ADC的长期稳定性评估仍可借鉴单抗与小分子药物开发积累的成熟技术体系。在质谱等尖端分析技术蓬勃发展的今天,需注意这些方法的阶段适用性。最终制剂开发阶段,稳定性指示方法必须具有长期一致性和稳健性,以支持ADC制剂表征所需的对比稳定性研究,并确保在产品生命周期可能涉及的多产地生产时具备方法可转移性。
ADC产品的整体开发高度依赖于其表征方法。本文概述了用于ADC原液分析表征的技术体系,重点阐释这些技术在ADC开发流程中的关键应用节点。我们将结合ADC生产工艺(图1.1,表1.3)及相关关键质量属性(表1.1),简要介绍质量控制(QC)工具的应用框架。
ADC表征的一个显著特点在于其活性分子固有的异质性——即使是最纯净的状态下,这种异质性也远超出起始单抗本身的翻译后修饰复杂性。目前已获批的ADC产品均存在连接子-载荷在个数和位点分布上的分布差异,这就要求从偶联阶段开始就必须对这种化学异质性进行系统表征和控制。多个连接子-载荷基团的共价连接会改变起始单抗的溶液性质,进而影响产品开发和表征策略。如表1.3所总结,ADC开发全过程必须进行的表征包括:化学稳定性、构象稳定性、胶体稳定性及总体溶液稳定性。
虽然现有多种分析技术可用于蛋白质溶液特性研究,但那些能反映ADC关键质量属性、并提供稳定性指示数据的检测方法才是ADC生产工艺的核心。表1.3同时标明了这些技术能解决的关键问题及其典型应用阶段。图1.2和图1.3则重点展示了在ADC产品生命周期中必须控制的多种外界作用力及降解途径。
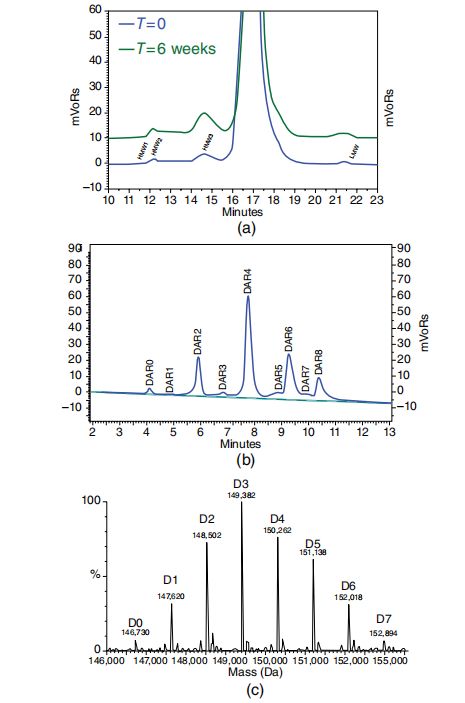
基于溶液的色谱与电泳技术是ADC开发中质量控制表征的核心手段。这些方法主要定量检测化学纯度与稳定性,也可部分反映构象稳定性。其中,尺寸排阻色谱(SEC)作为蛋白质纯化与分析的主要技术,能精准识别和定量由聚集形成的高分子量物质。结合多角度光散射(MALS)检测,SEC-MALS可监测ADC因载荷疏水性增加而导致的聚集倾向。即使低至1%的聚集水平(图1.4a)也能通过SEC检测,使其成为贯穿ADC生产流程的单抗稳定性重要指标。动态光散射(DLS)则能检测更大粒径的颗粒。
疏水作用色谱(HIC)在ADC表征中的应用日益广泛,该技术通过检测连接子-载荷数量增加导致的ADC分子疏水性变化(图1.4b),可在常规HPLC或UPLC系统上实现定性定量的DAR分布分析,为质量控制和稳定性评估提供关键数据。DAR值测定的正交方法紫外-可见光谱需依赖连接子或毒素上的合适发色团,但无法提供降解产物的分离信息。结合总蛋白分光光度检测,HIC与UV-vis应共同构成半胱氨酸偶联ADC的基础QC方案。而赖氨酸偶联ADC因异质性更高,需采用LC-MS技术解析更多组分。ADC的LC-MS分析通常需要去糖基化和变性条件,但SEC-MS联用技术可在非变性条件下进行质谱检测(图1.4c)。
成像毛细管等电聚焦(iCIEF)通过pH梯度凝胶分离,全程监测单抗与ADC的电荷分布。如图1.3所示,脱酰胺和赖氨酸截短(K缺失)等降解途径会改变分子电荷分布特征。还原/非还原SDS用于检测蛋白片段化。离子交换色谱(IEX)分析电荷态分布异质性。HIC、SEC和IEX等色谱方法可在非变性条件下进行,便于特定组分的分离制备或放大纯化。
反相HPLC主要用于细胞毒素和连接子衍生小分子的定量分析,常联用MS提升灵敏度。鉴于毒性风险,该分析对指导偶联后纯化至关重要,并能检测原液与制剂中游离毒素水平,通过识别多种潜在降解产物提供稳定性指示。其色谱条件取决于连接子/毒素特性,相关方法开发可借鉴连接子-药物的CMC数据。此外,因偶联中使用的DMSO/DMF等有机溶剂具有毒性,需通过气相色谱进行残留检测。
采用ELISA法检测ADC与起始单抗的抗原结合效价,可验证偶联过程是否导致亲和力下降。对于基于大肠杆菌或中国仓鼠卵巢(CHO)细胞表达系统的产品,已建立成熟的残留杂质定量方法:包括qPCR检测宿主细胞DNA、ELISA法检测宿主细胞蛋白等。这些单抗相关杂质必须在偶联前完成质量控制。
对于基于工程化半胱氨酸、非天然氨基酸或新型表达系统的下一代ADC分子,可能需重新优化上述检测方法。该原则同样适用于工程化抗体的血浆稳定性及药代动力学-药效学(PK-PD)特性评估——从表征角度而言,这类新一代单抗分子本身可能需被视为全新分子实体进行系统研究。
如同治疗性蛋白药物的开发,ADC原液或制剂也需进行深入表征,以提供尽可能全面的特性鉴定、纯度和稳定性信息,从而降低产品开发整体风险。为满足完整上市申报要求,需采用科学论证方法体系来确保原液和制剂在其市场生命周期内的特性一致性和可比性可控。部分检测方法虽不直接反映ADC稳定性,但能提供特性鉴定信息,例如氨基酸序列确证、连接子-药物位点占据率、单抗翻译后修饰特征以及偶联/纯化/制剂工艺引发的结构变化。反相高效液相色谱(RP-HPLC)肽图分析作为蛋白药物的常规指纹图谱技术,在ADC表征中占据重要地位。质谱技术是偶联活化和偶联步骤中评估异质性的关键手段。更全面的分子分析则需要采用LC-MS/MS等技术来确认氨基酸序列、单抗翻译后修饰及化学修饰,氧化、脱酰胺等复杂修饰及糖链特征的精准鉴定。
圆二色谱(CD)、紫外-可见光谱和荧光光谱等光谱技术正日益广泛地用于研究单抗构象/胶体稳定性及其与温度的相互作用。通过CD光谱可估算α螺旋和β折叠含量,既可对比初始单抗数据,也可通过长期监测进行稳定性评估。内源/外源荧光法已成功应用于化合物结合或制剂稳定性相关的二级/三级结构变化检测。差示扫描量热法(DSC)和动态光散射(DLS)等工具也正应用于ADC热力学研究。蛋白质构象完整性丧失或胶体不稳定的起始点受制剂稳定性影响。这些方法最适用于ADC原液纯化后监测连接子-载荷对单抗构象的影响,在制剂优化阶段则可用于筛选稳定辅料。此类方法提供的构象稳定性信息难以通过前述直接分子分析方法获得。需注意的是,这些生物物理方法在ADC表征和制剂优化中的普适性,很大程度上取决于所用连接子/载荷的光学特性、干扰因素及制剂组分。
与所有已上市原料药及制剂相同,必须证明ADC在整个生命周期内均能保持化学特性、药理学特性及毒理学特性的一致性。从监管角度而言,可比性研究的核心目的是确认生产工艺的微小变更不会对制剂的安全有效性产生负面影响。这种理化特性与功能的一致性必须贯穿于原材料供应商变更、生产场地变更及检测设备变更的全过程。实现可比性的前提是:首先在偶联工艺前建立单抗与药物/连接子材料的适当控制标准与验收标准。根据FDA要求,单抗及药物/连接子中间体需接受与终产品原料药同等水平的全面表征。对于单抗中间体,这包括完整的结构、纯度、稳定性及生物学活性评估,以确保当单抗生产发生变更时可进行可比性判定。同理,偶联用连接子/药物分子也需完成结构/元素分析、杂质谱分析及稳定性评估。随后通过偶联、纯化及制剂阶段的全面质量控制与深度表征数据,建立与历史批次或未来生产批次的可比性基础。可比性评估与标准制定的复杂性不容低估——从早期临床研究到产品上市期间将产生多个生产批次。ADC的可比性评估还存在额外复杂性:终产品由多个批次的单抗与连接子-药物中间体共同构成。鉴于特定分析结果与安全有效性之间的关联可能尚未完全阐明,ADC的可比性研究可能还需补充临床与非临床体内研究。
ADC的生产融合了细胞毒性小分子药物和单抗药物开发的诸多复杂性。总体而言,基于单抗的稳定性、偶联异质性以及这些关键质量属性(CQAs)的表征,是ADC生产工艺和优化开发方法的核心考量因素。ADC表征技术的发展速度与新一代ADC的创新步伐同步。预计随着偶联工艺的简化、ADC工艺认知的深入,以及阶段适配的表征工具的应用,未来将催生更安全、更有效且更具成本效益的治疗选择。